






The second year of the COVID-19 pandemic continued to dominate headlines, and the roll out of numerous vaccines in the U.S. and around the world promised to help slow the spread of the virus. Yet despite the disruptions to research and in-person medical care, areas of precision medicine made advances in 2021 which promise to improve patient care in the years ahead. Below are our picks for notable advances made over the past year.
Vaccinating adults against COVID-19
The first vaccines for COVID-19 were granted emergency use authorizations in the U.S. in December 2020, and the rollout of the Pfizer/BioNTech and Moderna vaccines (and later the single dose Janssen vaccine) featured heavily in the news cycle early in 2021. But while a pandemic-weary public had high hopes that the broad availability of vaccines would herald an end to the SARS-CoV-2-related restrictions imposed across the country, its availability couldn’t overcome resistance to receiving the vaccine based largely on political divisions within the U.S.—and within other countries around the world.
The vaccine rollout began in earnest at the beginning of the year, and within a couple of months all people aged 16 and older in the U.S. were eligible. But as we approach the end of 2021, still fewer than 60 percent of the U.S. population are fully vaccinated and a handful of states—predominantly in the Deep South and Intermountain West—have fewer than half of their residents vaccinated.
While vaccine skepticism and other factors have curtailed the number of people willing to roll up their sleeves, the publicizing of some adverse reactions from the Johnson & Johnson Janssen single-dose vaccine only provided fuel to those who were wary. In early spring, the FDA placed a pause on the use of the vaccine after reports of a small number of people experiencing rare blood clots and low platelet counts six to 13 days after receiving the vaccine. To date, a total of 28 people—more than three-quarter of them women—were reported to have experienced thrombosis with thrombocytopenia syndrome (TTS). TTS combines thrombosis involving the cerebral venous sinus thrombosis (CVST) and other sites in the body, including but not limited to the large blood vessels of the abdomen and the veins of the legs, along with thrombocytopenia or low blood platelet counts.
After reviewing the data from the Janssen vaccine, the FDA lifted the hold less than two weeks later. “We have concluded that the known and potential benefits of the Janssen COVID-19 Vaccine outweigh its known and potential risks in individuals 18 years of age and older,” noted FDA Commissioner Janet Woodcock, M.D., in a statement allowing vaccinations to resume. “We are confident that this vaccine continues to meet our standards for safety, effectiveness and quality.”
At the time, the agency noted only 15 TTS cases and three deaths had been reported from among 7.89 million who had received the vaccine. For this reason, some called the FDA to task for ever resorting to a well-publicized hold on use of the vaccine.
As Robert Nelsen, a co-founder and a managing director of venture capital firm ARCH Venture Partners, tweeted soon after the pause was lifted: ““JNJ fine. As expected. Damage done to world confidence in vaccines. Needless. Could have been a bulletin. Let’s move on but not do this again. There will be rare events again, but we are in a pandemic. Bulletins not pauses.”
First digital pathology diagnostic approved by FDA
Digital pathology has been breaking new ground, and at a rapid pace, the past few years. What began as a method for digitally storing images of stained tissue slides has now moved on to using artificial intelligence (AI) to quickly, via computing, identify or rule out the presence of cancerous cells. While digital pathology tools have shown the ability to more accurately identify cancer tissue than human pathologists, it has also become a vital research tool to help researchers better understand the heterogeneity of cancer, the tumor microenvironment, and single cell biological variations in cancer.
The technologies associated with digital pathology made a significant breakthrough in September when the FDA granted approval to Paige’s AI-driven Paige Prostate diagnostic—the first-ever digital pathology tool to gain U.S. regulatory approval as a diagnostic. While Paige Prostate had previously been CE-marked for use in hospitals in the European Economic Area, Switzerland, and the U.K., the regulatory approval in the U.S. has set the stage for a host of other digital pathology companies to spread their wings beyond its use as a research tool.
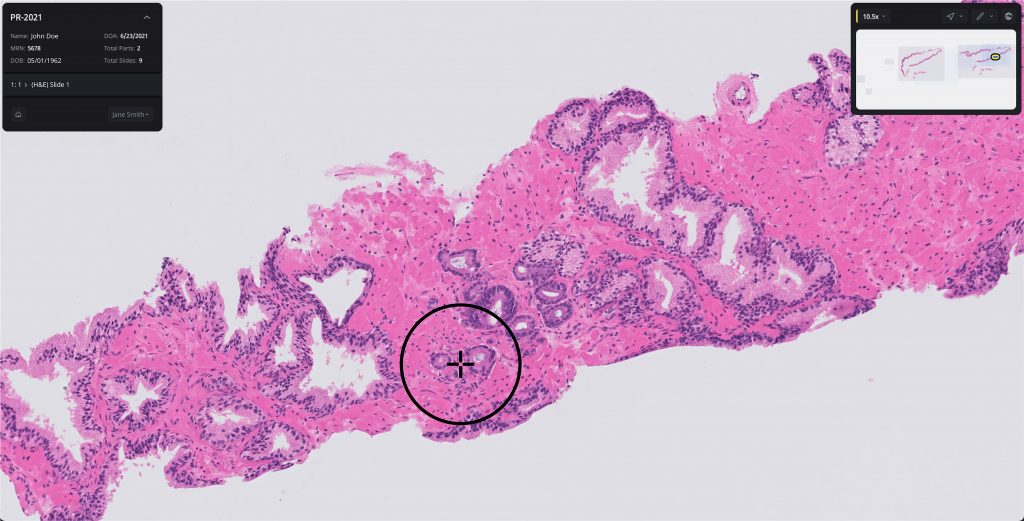
“The approval is a landmark achievement in the field of digital pathology and demonstrates how robust our technology is when faced with the broad range of natural variations in tissue slides encountered in day-to-day clinical practice,” Paige CEO Leo Grady, said at the time of the approval.
In the clinical study, 16 pathologists examined 527 slide images of prostate biopsies (171 cancer and 356 benign) that were digitized using a scanner. For each slide image, each pathologist completed two assessments, one without Paige Prostate’s assistance (unassisted read) and one with Paige Prostate’s assistance (assisted read).
While the clinical study did not evaluate the impact on final patient diagnosis, which is typically based on multiple biopsies, the study found that Paige Prostate improved detection of cancer on individual slide images by 7.3% on average when compared to pathologists’ unassisted reads for whole slide images of individual biopsies, with no impact on the read of benign slide images.
Pathologists using Paige Prostate also showed a 70% reduction of false negative diagnoses and a 24% reduction in false positive diagnoses. The dataset included slides from more than 150 institutions to ensure the system generalized to cases from different hospitals and different geographies.
“Pathologists examine biopsies of tissue suspected for diseases, such as prostate cancer, every day. Identifying areas of concern on the biopsy image can help pathologists make a diagnosis that informs the appropriate treatment,” said Tim Stenzel, M.D., Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health. “The authorization of this AI-based software can help increase the number of identified prostate biopsy samples with cancerous tissue, which can ultimately save lives.”
Whole-genome sequencing of critically ill babies proves its worth
When it comes to breaking new ground in the rapid sequencing of critically ill newborns in the neonatal intensive care unit (NICU), Rady Children’s Institute for Genomic Medicine (RCIGM) has few peers in delivering results for many of its patients. While it is safe to say that RCIGM has sufficiently proven its ability to identify the genetic variants driving many of these life-threatening rare conditions, it is now on a mission to bring its method of diagnosing sick babies to health systems across the country.

One of the hurdles to broader adoption is to not only show that its rapid Whole-Genome Sequencing (rWGS) method not only helps NICU physicians save babies’ lives, but also that implementing a program to do so saves money versus other methods—a key measure to get both public and private healthcare payers to reimburse for this service.
In June, RCIGM delivered early data from its collaboration with Medi-Cal, California’s Medicaid program, that showed its whole-genome sequencing not only improved health outcomes overall for these sick babies, but also saved that state insurance program more than $2.5 million in costs over two years.
Dubbed Project Baby Bear at its launch in 2018, the pilot project with California became the first state funded program to use whole-genome sequencing as a method of care for critically ill newborns who were suspected of having a rare genetic disease.

At the time of the pilot’s launch, Stephen Kingsmore, M.D., president and CEO of RCIGM noted: “we’ve seen that using whole genome sequencing to diagnose and guide the care of babies hospitalized with rare diseases is reducing suffering and infant mortality, decreasing hospital stays and healthcare costs. It’s our belief that rapid whole genome sequencing should become a first-line diagnostic test and standard of care in neonatal intensive care units everywhere.”
Data from Project Baby Bear demonstrated this. Using rWGS proved a valuable diagnostic tool for clinicians and helped diagnose rare genetic diseases that explained the baby’s admission to intensive care in 74 babies or 40% of the 184 babies in the Medi-Cal program. Of the babies diagnosed using this technology, 58 (32%) had information uncovered from sequencing that allowed physicians to take a precision medicine approach to their care.
Such early treatment for these babies also helped reduce unnecessary treatments such as tracheostomies or gastric tube insertions, eliminated the need for further testing and led to fewer days in the hospital—all of which resulted in lower costs of caring for the critically ill newborn population.
“It’s very rare to be able to use the most advanced medical technology to both improve a child’s life and save money, yet Project Baby Bear proved it’s possible,” noted David Dimmock, M.D., senior medical director of RCIGM. “We identified the exact cause of rare, genetic diseases in an average of three days, instead of the four to six weeks standard genetic testing offers. This allowed physicians to deliver timely treatment tailored to the baby’s specific condition, in some cases avoiding irreversible health consequences.”

The ripple effects of this groundbreaking pilot are already in evidence.
Even before RCIGM released the Project Baby Bear data mid-year, Blue Shield of California announced in June of 2020 that it would cover rWGS to help diagnose critically ill children in intensive care with unexplained medical conditions. Michigan, Florida, and New Jersey have all rolled out their own publicly funded versions of the model using rWGS for critically ill babies. And, in late June, U.S. Senators Susan Collins (R-ME), Mark Kelly (D-AZ), and Bob Menendez (D-NJ) introduced S. 2022 the “Ending the Diagnostic Odyssey Act” that would provide funding to allow all 50 states to conduct whole-genome sequencing of children in the Medicaid program suspected of having a rare genetic disease. The bill proposes a three-year pilot program, under which the federal medical assistance percentage (FMAP) would be 75% of the total costs, with states picking up the remainder.
“As the mother of two kids who experienced the diagnostic odyssey, I know the anguish felt by parents as they wait for a diagnosis,” said Sharon Terry, president and CEO of the Genetic Alliance, in a press release announcing the legislation. “Thanks to advances in genomic sequencing, we now can diagnose rare genetic diseases in days, instead of months or years, saving lives and saving money.”
Comprehensive cancer genomic profiling picks up steam
While cancer has long been on the cutting edge of the development of precision medicine approaches to treating disease, the early years of targeting the genetics of cancer development, whether driven by germline or somatic mutations, tended to employ smaller gene panels for cancer testing. But today, aided by the reduction in costs of whole-genome sequencing, physicians and diagnostics companies are increasingly turning to a new tool—comprehensive genomic profiling, or CGP.
With CGP, clinicians can obtain genomic information about a patient’s cancer focused on not just a few dozen genes, but on hundreds of genes in single test. Typically, these tests detect base substitutions, insertions and deletions, copy number alterations (CNAs), and rearrangements or fusions—the four different kinds of genomic alterations that are known to be the drivers of cancer development and growth.
The concept behind running a single test on a patient’s tissue sample or from a liquid biopsy is simple: running panels with more than 300 genes in a single test will speed the time to diagnosis, allowing physicians to select targeted therapies most appropriate for each patient’s unique molecular profile.

“We have now an incredible number of very powerful molecular based therapies for oncology patients and there is a lot of hope patients in the years to come to have more and more options that will translate to better clinical outcomes,” Luca Quagliata, vice president, global head of medical affairs at Thermo Fisher Scientific, told Clinical OMICs, early in 2021. “But there is one barrier in between having these treatments and getting to these treatments and that is testing. No testing, no treatment. It is pretty straightforward.”
In addition, CGP may become standard of care in the coming years as it can providing valuable information used for sniffing out cancer recurrence. A host of companies are currently racing to develop liquid biopsies that can detect cancer recurrence months before traditional imaging techniques. The concept is that liquid biopsies can be performed on an ongoing basis for patients in remission to get the earliest jump on treating metastatic cancer. And using the information from CGP performed at first diagnosis can inform which genetic alterations to look for in individual patients should their cancer return. That means liquid biopsy test developers can create a streamlined, custom testing panel for each patient that searches only for the drivers of their cancer. Further, as these recurrent tests would typically leverage PCR as the testing platform, costs can remain in check making them an attractive option for recurrent screening.

One of the issues in the U.S. that may slow the use of CGP is whether public and private payers will reimburse for the tests, which are more expensive than panels with fewer genes. To help advance the evidence for payers—and accelerate CGP adoption—lab services and diagnostic developers came together in late 2020 to create the Access to Comprehensive Genomic Profiling Coalition (ACGP), whose mission is to advocate for the effectiveness of CGP in advanced cancer care and for broader coverage of these tests with both public and private payers.
Chair of ACGP Jim Almas, M.D., vice president and national medical director of clinical effectiveness at LabCorp told Clinical OMICs: “The idea is that patients will get better if they have better access to these tests. It is also critical they have access to these tests for better opportunities to enroll in clinical trials.”






