






By Donna Snyder
One of the quandaries that researchers face when considering a clinical study in children is balancing the need to protect this vulnerable patient population with the need to conduct pediatric research that can improve their health and well-being.
The ethical framework for pediatric research was developed to help guide those discussions. It states that children should only be enrolled in a clinical trial or in research if scientific or public health objectives cannot be met by enrolling participants who can consent personally. When possible, data from adults should be extrapolated to children to minimize the need to collect data in children. Furthermore, children should not be placed at a disadvantage by being enrolled in a clinical trial either through exposure to excessive risks or by failing to get necessary healthcare. If there is no prospect of a clinical benefit, the risks to which children are exposed must be low. Also, vulnerable populations, including children, who are unable to consent must have a suitable proxy to consent for them.
FDA pediatric guidance
The United States Food and Drug Administration (FDA) regulations for human subject protection in clinical research are found at 21 CFR 50 and 21 CFR 56, and for the Department of Health and Human Services (HHS) are found at 45 CFR 46. Both these regulations include special protections for children. They are nearly identical except for some minor differences, found in 21 CFR 50, subpart D and 45 CFR 46, subpart D, respectively.

The FDA has also developed draft guidance specifically for FDA-regulated pediatric research: “Ethical Considerations for Clinical Investigations of Medical Products Involving Children” was issued in September 2022, together with a supporting document or “snapshot” and a podcast that highlights key points.
As noted above, ethical pediatric research is guided by several important principles.
Principle of scientific necessity
This states that children should not be enrolled in a clinical trial unless it is necessary to answer important scientific or public health questions related to children. Furthermore, subjects capable of informed consent (usually adults) should generally be enrolled prior to children (21 CFR 56.111(a)(3)), and any research procedures that do not contribute to a scientific objective should be eliminated (21 CFR 56.111(a)(1)).
When this framework was being developed, the National Commission was considering drugs that were being developed in adults and generating data that could be applied to children. But now, some rare disease researchers are focusing on conditions that may not exist in adults. In such cases, scientific necessity may require that these studies start with children and may never include adults.

A classic example is nusinersen (Spinraza®), a drug approved by the FDA to treat spinal muscular atrophy. The studies were performed with pediatric participants, but the product approval was extended to adults. The studies were initiated in children because children are more severely affected by the disease and were most likely to benefit from the treatment. Additionally, because the disease is less severe in adults, an efficacy signal might not have been seen in adults without a very large study.
Pediatric extrapolation
This concept allows efficacy data to be extrapolated from well controlled adult studies to children in instances when the course of the disease and effects of the drug are similar in both populations.4 The efficacy data is usually supplemented with dosing information obtained from pediatric patients.
This concept can also be used to extrapolate efficacy data from one pediatric subpopulation to another, such as from adolescents to younger children, and can help streamline a pediatric drug development program.
Justifying research risk
For adults, Institutional Review Board (IRB) approval of research is generally justified if the risks to the participants are reasonable in relation to the anticipated benefits, if any, to participants and the importance of the knowledge that may be expected to result (21 CFR 56.111(a)(2)). The IRB assesses the risk:benefit balance and determines whether it is acceptable. With children, this criterion is modified because there is a limit to the risk that knowledge alone can justify.
Research involving children either must be restricted to minimal risk or a minor increase over minimal risk if there is no potential for direct benefit to the enrolled child (21 CFR 50.51/3) or must present risks that are justified by the prospect of direct benefit to the child; the balance of which is at least as favorable as any available alternatives (21 CFR 50.52).
There are some instances when an IRB determines that it cannot approve a study under the previous criteria, but the research may present an opportunity to understand, prevent, or alleviate a serious problem affecting the health or welfare of children. On those occasions, the IRB can refer the protocol to the FDA, or the Office for Human Research Protections (OHRP) if it is federally funded, for review by a federal panel (21 CFR 50.54). The public can also provide feedback on the submitted protocol. The panel makes a recommendation to the FDA Commissioner, or in the case of an OHRP review to the Secretary of HHS, who then decides whether the study should move forward. For studies that are federally funded and include an FDA-regulated product, a joint review by FDA and OHRP will occur.
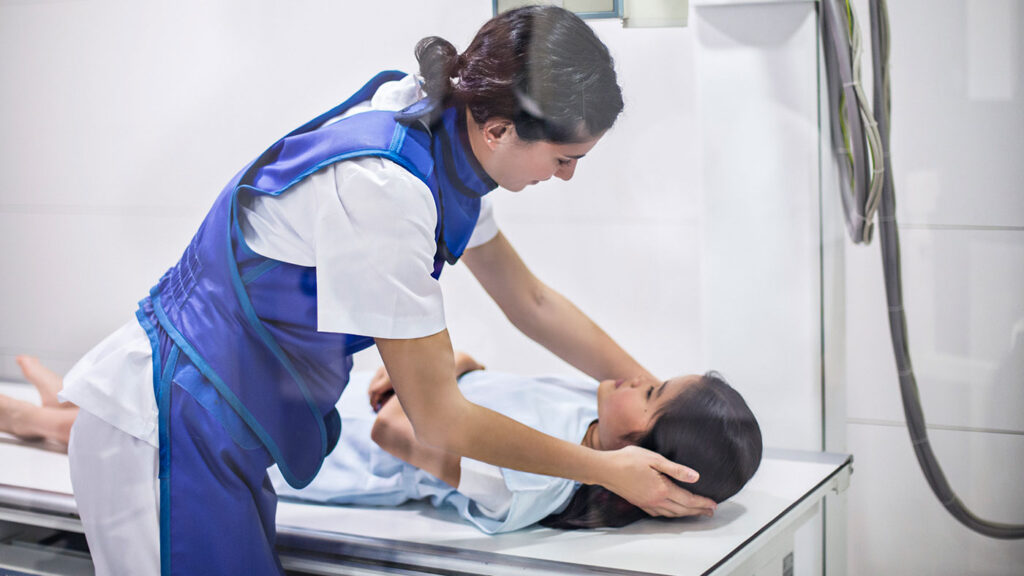
Minimal risk
Minimal risk is defined in the regulations as the “probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests” (21 CFR 56.102(i)). This criterion relates to a healthy child.
Procedures that are considered minimal risk include a single blood draw, a physical exam, a chest x-ray, or surveys. An investigational drug used in a clinical investigation that includes children is generally not considered minimal risk. A device used in a clinical investigation could be considered minimal risk depending on how it is used.
Minor increase over minimal risk
The National Commission recognized there would likely be occasions, particularly with therapeutic research, where it would not be possible to categorize research as either minimal risk or offering a prospect of direct benefit. For example, there could be situations where it might be helpful to collect data in children, but the data may only contribute to generalizable knowledge and the risk might be greater than minimal. Therefore, it developed the “minor increase over minimal risk category,” which is defined as risk that goes slightly beyond the narrow boundaries of minimal risk but poses no significant threat to the child’s health or well-being. However, it can only be applied to children who have, or are at risk of getting, the disease or condition about which the study could yield generalizable knowledge.
Procedures that are considered a minor increase over minimal risk include collecting urine via a catheter, a single lumbar puncture, or a single dose of an investigational drug with adequate safety information. For example, a single-dose pharmacokinetics study in a pediatric population could qualify if all the other procedures in the clinical study were either minimal risk or no more than a minor increase over minimal risk.
Component analysis
When an IRB reviews a pediatric research protocol, it conducts a component analysis of the research-related interventions or procedures to ensure that participants will not be exposed to unacceptable risks without benefit.
In addition to the study drug, biologic or device which is expected to offer the prospect of direct benefit, a clinical trial may require additional procedures or interventions, for example, magnetic resonance imaging (MRI) or a lumbar puncture, which do not necessarily benefit the child, but may be needed to assess some of the research outcomes.
Each intervention that is conducted solely for research purposes, and not needed for clinical management or routine clinical care, must be evaluated separately to determine whether it offers the prospect of direct benefit to the enrolled child. If it does not, then that intervention or procedure should be limited to minimal risk or a minor increase over minimal risk, and meet the other conditions outlined under 21 CFR 50.51 or 21 CFR 50.53, respectively.
Placebo-controlled trials
For children enrolled in the active study arm, there is a prospect of direct benefit, and the risk of administration can be balanced by the potential benefit of receiving the medical product. But for children in the placebo arm, there is no prospect of direct benefit, so the risks should be minimal or no more than a minor increase over minimal risk.
When determining whether a placebo poses no more than a minor increase over minimal risk, the IRB must consider the intervention (e.g., sugar pill or saline), route of administration (e.g., oral, infusion, topical) and any procedures used for administration (e.g., placement of a peripheral catheter). It must also factor in the frequency and duration of administration, the risk of withholding known effective therapy (if one exists and would be withheld), and the use of rescue therapy, if appropriate.
If the protocol requires a child to receive sedation for a non-therapeutic procedure, such as an MRI, then the risks of sedation must be evaluated by the IRB. If the procedure is part of clinical care and offers a clinical benefit to the child, then it does not need to be assessed as a research procedure. Sedation or imaging studies that are required for placement of a device that could provide direct benefit would be considered under 21 CFR 50.52.
Biopsies require special consideration. For example, muscle biopsies, with the associated anesthesia, that are used for patients with Duchenne muscular dystrophy have been determined by IRB precedent to meet the minor increase over minimal risk criterion. But large organ biopsies, such as those of the liver or kidney, exceed that threshold. Therefore, they are only included in a protocol if needed for clinical care.
When evaluating diagnostic imaging studies, the IRB must consider the total radiation exposure and type of contrast used. Participants’ radiation exposure should be limited to that needed to meet the studies’ scientific objectives and to evaluate study outcomes.

Defining “children”
Under the regulations (21 CFR 50, subpart D), children are defined as “persons who have not attained the legal age for consent to treatments or procedures involved in clinical investigations, under the applicable law of the jurisdiction in which the clinical investigation will be conducted,” i.e., where the study site is located (21 CFR 50.3(o)). In most states, that is age 18, but there are a couple of states where it is older.
But subpart D may not apply to minors who have the legal right to consent for certain treatments or procedures included in the clinical investigation. For example, adolescents in some states can consent to be treated for sexually transmitted diseases or obtain advice regarding reproductive health issues without their parents being informed. If they can consent for themselves for those procedures, they can also consent as an adult in the research study.
Informed consent
The child’s parent/guardian must give permission via the informed consent process and the child must give affirmative agreement or assent (if they are able to provide it) before the child can be enrolled in a study (21 CFR 50, subpart B, 21 CFR 50.3(r) and 21 CFR 50.55).
In most cases, children aged seven or older are considered able to assent to participate in research. But the IRB may adjust the age requirement based on the maturity and psychological state of the patient population. As the study progresses and new information is learned, the parent/guardian and child should also be asked whether they want to continue participating.
For informed consent to occur, the parental permission form must provide sufficient information for the parent/guardian to be able to make an informed decision and they must also be given the opportunity to ask questions.
Conclusion
Involving pediatric patients in clinical studies necessitates striking a delicate balance between protecting vulnerable patients while still allowing them to participate in scientific research which could yield new treatments that could improve their health and quality of life. To that end, the FDA and HHS have established a regulatory and ethical framework to guide the IRB review of pediatric research protocols and help ensure that the potential benefits outweigh any risks involved.
Read More:
- E11A Pediatric Extrapolation, FDA Guidance, August 2022
- Additional Safeguards for Children in Clinical Investigations, 21 CFR 50, subpart D
- Additional Protections for Children Involved as Subjects in Research
- Ethical Considerations for Clinical Investigations of Medical Products Involving Children, FDA, Draft Guidance, September 2022, Snapshot Podcast
- Research Involving Children as Subjects and Not Otherwise Approvable by an Institutional Review Board: Process for Referrals to Food and Drug Administration and Office for Human Research Protections E11A Pediatric Extrapolation, FDA Draft Guidance, March 2023
- Research Involving Children, September 1977
- General Clinical Pharmacology Considerations for Pediatric Studies of Drugs, Including Biological Products, FDA Draft Guidance, September 2022
- Informed Consent Guidance for IRBs, Clinical Investigators, and Sponsors,
August 2023
Donna Snyder, MD, MBE, is executive physician at WCG providing guidance on medicine, research, and regulations. Before WCG, Snyder served as senior pediatric ethicist, in the Office of Pediatric Therapeutics, at the U.S. FDA. She is a board-certified pediatrician with experience in pediatric practice and research ethics.






